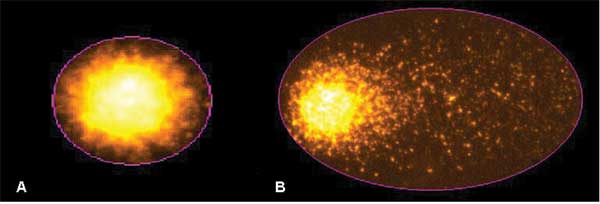
Figura 1 - Avaliaçăo dos níveis de estresse oxidativo (“Tail
Moment”), por meio da eletroforese em gel de célula única (ensaio
do cometa). A – Célula normal; B – Célula com dano oxidativo
intenso.
ARTIGOS ORIGINAIS
AVALIAÇĂO DA EXPRESSĂO TECIDUAL DO GENE DE REPARO MLH1 E DOS NÍVEIS DE DANO OXIDATIVO AO DNA EM DOENTES COM CÂNCER COLORRETAL
Evaluation of Expression of Mismatch Repair Gene MLH1 and Levels of Oxidative DNA Damage in Normal and Neoplastic Tissues of Patients with Colorectal Cancer
Carlos Augusto Real Martinez1, Adriana Teixeira Cordeiro2, Denise GonÇalves Priolli2; Daniel Duarte da ConceiÇÃo Miranda2, Waldemar Bartchewsky JÚnior3, Nelson Fontana Margarido4, Marcelo Lima Ribeiro5
1 Professor Adjunto do Programa de Pós-Graduação em Ciências da Saúde da Universidade São Francisco. Chefe do Serviço de Cirurgia Geral do Hospital Universitário São Francisco, Bragança Paulista, (SP); 2 Acadêmicos do Curso de Medicina da Universidade São Francisco, Bragança Paulista, (SP). Bolsistas e Iniciação Científica (FAPESP); 3 Pós-Graduando em Ciências da Saúde da Universidade Federal de São Paulo, (SP); 4 Professor Livre Docente do Departamento de Cirurgia da Faculdade de Medicina da Universidade de São Paulo, (SP); 5 Professor Assistente do Programa de Pós-Graduação em Ciências da Saúde da Universidade São Francisco, Bragança Paulista, (SP).
Resumo: O dano oxidativo ao DNA provocado por radicais livres de oxigênio representa um dos principais mecanismos responsáveis pelas etapas iniciais da carcinogênese colorretal. O estresse oxidativo ocasiona erros de pareamento de bases possibilitando o aparecimento de mutações em genes controladores do ciclo celular. As células possuem um sistema de defesa representado pelos genes de reparo do DNA que corrigindo os erros de pareamento impedem o desenvolvimento de mutações. Poucos estudos avaliaram a relação entre dano oxidativo ao DNA e a expressão tecidual do gene de reparo MLH1. Objetivo: O objetivo do presente estudo foi avaliar os níveis de estresse oxidativo ao DNA e a expressão tecidual do gene de reparo MLH1 nas células da mucosa cólica normal e neoplásica de doentes com câncer colorretal. Material e Método: Foram estudados 44 doentes com diagnóstico de adenocarcinoma colorretal. Foram excluídos os doentes com câncer colorretal hereditário, portadores de câncer relacionado às doenças inflamatórias intestinais e os submetidos à radioquimioterapia neoadjuvante. Para a avaliação dos níveis de dano oxidativo ao DNA utilizou-se a técnica da eletroforese alcalina em gel de célula isolada (ensaio do cometa) avaliando 100 células obtidas dos tecidos normal e neoplásico. Para a avaliação da expressão do gene MLH1 utilizou-se a técnica de reação de polimerase em cadeia em tempo real (RT-PCR) com primer especificamente desenhados para amplificação do gene. A comparação dos resultados encontrados para os níveis de estresse oxidativo ao DNA, e expressão do gene MLH1 nos tecidos normais e neoplásicos foi feito pelo teste t de Student, adotando-se nível de significância de 5% (p<0,05). Resultados: Os níveis de dano oxidativo ao DNA no tecido neoplásico foram significativamente mais elevados quando comparados ao tecido normal (p=0,0001). A expressão tecidual do gene MLH1 no tecido neoplásico foi significativamente menor quando comparado ao tecido normal (p=0,02). Conclusão: O gene de reparo MLH1 encontra-se menos expresso no tecido neoplásico e inversamente relacionado aos níveis de dano oxidativo ao DNA.
Descritores: Câncer Colorretal, Dano do DNA, Reparo do DNA, Estresse oxidativo, Ensaio em Cometa, Reaçăo em Cadeia da Polimerase.
INTRODUÇÃO
O câncer colorretal (CCR) é a terceira neoplasia maligna mais frequente que acomete o homem, representando uma das principais causas de morte em todo
mundo.1,2 Não obstante os recentes avanços obtidos no diagnóstico precoce e tratamento, os índices de mortalidade nas últimas décadas pouco se alteraram1. No Brasil, a evolução do CCR tem apresentado comportamento semelhante sendo, atualmente, a quarta causa mais comum de morte relacionada ao câncer e, a exemplo do que acontece em outros países, sua incidência vem aumentando em comparação a outros tipos de tumores do aparelho digestivo.2 Estimativas mostram que no Brasil em 2008, ocorreram 27.000 casos novos de CCR, correspondendo a um risco estimado de 13-15casos-novos/100.000 habitantes.2 O aumento da expectativa de vida, os fenômenos da globalização e, principalmente, a mudança de hábitos dietéticos fizeram com que o CCR ganhasse importância crescente no perfil da mortalidade por câncer em todo o mundo.1
Desde a publicação do trabalho pioneiro de Fearon e Vogelstein em 19903, encontra-se bem estabelecido que o surgimento do CCR, a partir da mucosa cólica normal, é mediado por uma sequência de mutações em genes controladores do ciclo celular (proliferação, diferenciação, e apoptose) ou em genes responsáveis pelo reparo do DNA. A partir de então os grandes avanços no campo da biologia molecular vem permitindo a melhor compreensão dos mecanismos genéticos e moleculares envolvidos na Scarcinogênese colorretal.4
O desenvolvimento do CCR é
essencialmente um processo evolutivo somático clonal, envolvendo
uma série de mutações ou mudanças na expressão de
vários genes. Essas alterações conferem vantagens
adicionais para o crescimento tumoral, em relação ao
tecido normal.4 Contudo, os mecanismos iniciais
responsáveis pela transformação da célula normal em
célula neoplásica, ainda não se encontram totalmente
esclarecidos.4 Estudos demonstraram que o dano ao
DNA provocado pelos radicais livres de oxigênio (RLO) e
a hipermetilação, incorporação de radicais metil
(CH3) na região promotora dos genes, representam dois
dos principais mecanismos relacionados às etapas
iniciais do desenvolvimento do CCR.5, 6,7,8,9,10,11,12,13 ,14,15
Os RLO são constantemente formados
durante o metabolismo energético normal de todas as
células vivas.14 Entretanto, quando sua produção é
excessiva, tornam-se nocivos às células danificando
proteínas, membranas, organelas e bases nitrogenadas do
DNA.16 Para se defender as células possuem sistemas de
defesa antioxidantes, enzimáticos e não-enzimáticos,
que atuam contra a toxicidade dos RLO sendo
responsáveis pela manutenção da homeostase entre produção
e
neutralização.14,15,16,17 Em certas condições, quer
pela diminuição dos sistemas antioxidantes do
organismo, quer pelo aumento exagerado na produção de
RLO, ocorre desequilíbrio, determinando o aparecimento
de fenômeno conhecido como estresse
oxidativo.16,17,18,19 Da mesma forma, a produção excessiva de
radicais CH3 podem ocasionar hipermetilação da
bases nitrogenadas que compõe as regiões promotoras
dos genes determinando o bloqueio de sua transcrição.
Estudos demonstraram que o estresse oxidativo provoca quebras simples e duplas nas fitas que
compõem as moléculas do DNA e induz a erros
de pareamento das bases nitrogenadas
possibilitando, consequentemente, aparecimento de mutações
genéticas.19,20,21,22 Quando a mutação ocorre em genes
responsáveis pelo controle do binômio
proliferação/morte celular poderá haver formação de um clone de
células com autonomia proliferativa ou resistência a
apoptose, características inerentes a célula
neoplásica.23 Para que isso não ocorra, as células possuem
mecanismos de defesa contra esses erros de pareamento
representados pelos sistemas de reparo do DNA
(mismatch repair-
MMR).6,7,8,9,23 Os genes que compõe o
sistema de reparo traduzem proteínas cuja função
primordial é corrigir os erros de pareamento de bases
impedindo a transmissão destas mutações a futuras
gerações celulares.23 Dentre os principais genes de
reparo destacam-se o MLH1, MSH2, PMS1, PMS2, MSH6 que apresentam importância na correção de
mutações relacionadas ao estresse oxidativo e ao MGMT
com importância na correção da incorporação de
radicais CH3 em bases do DNA. 6,7,8,9,24,25,26
Recentes estudos vêm demonstrando que as células oriundas de tecidos neoplásicos obtidos de
doentes com CCR apresentam maiores níveis de
dano oxidativo ao DNA.22,27 Todavia, também
demonstraram que no tecido normal desses mesmos
doentes, apesar de existirem níveis consideráveis de
dano oxidativo ao DNA as células não apresentam
transformação
neoplásica.20,22,27. É provável que a
neoplasia não se desenvolva pela ação das proteínas de
reparo que corrigem os danos ao DNA, provocados pelos
RLO e pela hipermetilação. É possível que no
tecido neoplásico, o sistema de reparo não consiga
corrigir, com a mesma eficiência, os erros de pareamento
de bases permitindo o surgimento de células com
mutações genéticas. Essa possibilidade torna atraente o
estudo das relações existentes entre estresse oxidativo
e capacidade de reparo do DNA, comparando tecidos normais e neoplásicos.
O objetivo do presente estudo foi avaliar a expressão do gene MLH1 e os níveis de estresse
oxidativo ao DNA nuclear comparando células normais
e neoplásicas da mucosa cólica de doentes com CCR.
MATERIAL E MÉTODO
O presente estudo recebeu aprovação do
Comitê de Ética em Pesquisa da Universidade São
Francisco. Todos os pacientes que forneceram material
biológico para a pesquisa assinaram Termo de
Consentimento Livre e Esclarecido após serem informados
de todas as etapas experimentais.
Casuística
Foram elegíveis para o presente estudo 44
enfermos (22 mulheres), com média de idade de 62,4
anos, portadores de adenocarcinoma colorretal,
submetidos a tratamento cirúrgico com intenção curativa, por
uma mesma equipe cirúrgica entre janeiro de 2007 e
dezembro de 2008. Foram excluídos os doentes com
CCR hereditário (polipose adenomatosa familiar e
câncer colorretal hereditário não polipóide), portadores de
CCR associado à doença inflamatória intestinal,
doentes operados em caráter de urgência e, aqueles
submetidos a tratamento radioterápico ou
quimioterápico neoadjuvante.
Coleta do material
Imediatamente após a remoção do
espécime cirúrgico, foram retirados seis fragmentos da
mucosa cólica normal distante no mínimo 10 cm da
margem superior da neoplasia. Da mesma forma, foram
colhidos seis fragmentos da mucosa neoplásica obtidos
da periferia do tumor. Os fragmentos foram
identificados e acondicionados, individualmente, em recipientes
apropriados, e imediatamente, enviados ao Laboratório
de Biologia Molecular da Universidade São Francisco,
onde foram resfriados a _80ºC, até o momento da
realização das análises moleculares. Três fragmentos de
cada tecido (normal e neoplásico) foram destinados ao
estudo da expressão do gene MLH1 pela reação em
cadeia de polimerase em tempo real (RT-PCR). Os
seis fragmentos restantes provenientes dos tecidos
normais e neoplásicos foram acondicionados em recipiente
com solução de congelamento e destinados a
mensuração dos níveis de estresse oxidativo ao DNA pela
técnica da eletroforese alcalina em gel de célula isolada
(ensaio do cometa). Os ensaios laboratoriais foram
realizados em um único momento. Todos os doentes
selecionados foram diagnosticados como portadores
de adenocarcinoma pela técnica da
hematoxilina-eosina por patologista experiente em neoplasias
colorretais, que desconhecia os objetivos do presente estudo
Eletroforese alcalina em gel de célula isolada (ensaio do cometa)
Para a realização do ensaio do cometa
foram utilizados os três fragmentos obtidos do
tecido neoplásico e três fragmentos de tecido normal. A
análise dos danos oxidativos ao DNA das células da
mucosa cólica foi feita de acordo com método
anteriormente proposto.28 Resumidamente, as amostras foram
incubadas em 3ml de uma solução de Hank's
(HBSS, Invitrogen, Carlsbad, CA, USA) contendo 5,5 mg
de proteinase K (Sigma Chemical CO, St. Louis, MO) e
3 mg de colagenase III (InvitrogenÒ) por 45 minutos
a 37ºC para a liberação das células. A seguir, foram
re-suspendidas em 10 ml de HBSS e centrifugadas para
o isolamento das células. Realizamos o ensaio do
cometa apenas nas amostras que apresentassem
viabilidade celular maior que 75%. Avaliamos a viabilidade
celular usando o método do diacetato de fluoresceína (FDA)
/ brometo de etídio (EtBr- Sigma-Aldrich,
St Louis,MO,USA). Para o estudo da viabilidade, a
solução de coloração celular foi preparada
imediatamente antes da sua utilização e continha 30 ml de FDA
em acetona (5mg/ml), 200 ml de EtBr em tampão
fosfato (PBS; 200 mg/ml), e 4,8 ml de PBS
(Invitrogen, Carlsbad, CA, USA). A suspensão contendo
células isoladas foi então misturada com 25 ml da
solução corante, colocada sobre lâmina e recoberta
com lamínula. As lâminas foram lidas em microscópio
de imunofluorescência. Ao núcleo das células viáveis
coravam-se em verde, enquanto das células inviáveis
em vermelho. Após análise das lâminas,
selecionamos amostras dos tecidos que apresentassem mais de
75% das células viáveis.
A versão alcalina do ensaio do cometa foi
realizada de acordo com Ladeira et al.,
2004.29 De forma resumida, 15 ml da suspensão celular previamente
obtida foram misturadas a agarose "low melting
point" 0,5 % (Promega), postos sobre uma lâmina e
cobertos com uma lamínula. Em seguida foram imersas em
uma solução de lise gelada (2,5 M NaCl, 100 mM
EDTA, 10mM Tris, 1% SDS, pH 10 com 1% Triton X-100
e 10% DMSO) permanecendo a 4ºC por 12 horas. Subsequentemente, foram expostas a um tampão
alcalino (1 mM EDTA e 300 mM NaOH, pH ~13,4) por 40 min a 4ºC. A eletroforese foi realizada neste
tampão a 4ºC por 30 min a 25V e 300 mA. Após a
realização da eletroforese, as lâminas foram neutralizadas
(0,4 M Tris, pH 7,5), coradas com SYBR Safe
(Invitrogen, Carlsbad, CA, USA) e analisadas com um
microscópio de fluorescência. Duzentas células foram
aleatoriamente selecionadas (100 de cada amostra tecidual)
e analisadas individualmente utilizando-se o
software Komet 5.5 (Kinetic Imaging Ltd. _ USA).
Com o auxílio do software obteve-se o
valor da extensão da cauda do cometa (Tail
Moment) sendo seus valores médios determinados. Segundo o
manual do fabricante, o Tail Moment é definido como o
produto do DNA da cauda e a distância média da
migração da cauda. O tamanho da cauda reflete a extensão
das rupturas das hélices de DNA (dano oxidativo), e
pode ser quantificado por métodos de intensificação de
imagem (Figura 1). Adotou-se como valor final para
os níveis de dano oxidativo ao DNA nos tecidos normal
e neoplásico a média dos valores encontrados nas
três amostras analisadas de cada tecido.
|
Figura 1 - Avaliaçăo dos níveis de estresse oxidativo (“Tail
Moment”), por meio da eletroforese em gel de célula única (ensaio
do cometa). A – Célula normal; B – Célula com dano oxidativo
intenso. |
 |
 |
 |
ABSTRACT: The oxidative DNA damage caused by oxygen free radicals is one of the most important mechanisms responsible for the initial steps of colorectal carcinogenesis. The oxidative stress can cause errors in the pairing of nitrogenous bases that form the DNA, allowing mutations in controlling genes of the cell cycle. The cells have a defense system represented by the DNA mismatch repair genes that correct the errors of matching prevent the development of DNA mutations. Few studies have evaluated the relationship between oxidative DNA damage and the tissue expression of mismatch repair genes. Aim: The aim of the present study was evaluate the levels of oxidative DNA and the tissue expression of MLH1 mismatch repair gene in the cells of normal and neoplastic colonic mucosa of patients with colorectal cancer. Material and Methods: Were studied 44 patients with diagnosis of colorectal adenocarcinoma. Were excluded patients with hereditary colorectal cancer, with colorectal cancer associate with inflammatory bowel diseases and those undergoing neoadjuvant radioquimiotherapy. To evaluate the levels of oxidative DNA damage was used the single cell gel electrophoresis (comet assay) evaluating 100 cells obtained from normal and neoplastic tissues. For the evaluation of the tissue expression of MLH1 gene was employed the technique of polymerase chain reaction in real time (RT-PCR) with primer specifically designed for MLH1 gene. The comparison among the levels of DNA oxidative stress and expression of MLH1 mismatch repair gene in normal and neoplastic tissues was done by Student t test adopting a significance level of 5% (p< 0.05). Results: The levels of oxidative DNA damage in tumor tissue were significantly higher when compared to the level of the normal tissue (p = 0.0001). The tissue expression of MLH1 mismatch repair gene in tumor tissue was significantly lower when compared to normal tissue (p=0.02). Conclusion: The mismatch repair gene MLH1 are less expressed in tumor tissue and inversely related to levels of oxidative DNA damage.
Key words: Colorectal Cancer, DNA Damage, DNA Repair, Oxidative stress, Comet Assay, Polymerase Chain Reaction.
Referências
1. Boring CC, Squires TS, Tong T, Montgomery S.
Cancer statistics 1994. CA Cancer J Clin.1994;44(1):7-26.
2. Instituto Nacional do Câncer. Estimativa 2008. Incidência
de câncer no Brasil. Disponível em URL:
http://www.inca.gov.br/estimativa/2008/versaofinal.pdf Acesso em: 20/06/2009.
3. Fearon ER, Vogelstein B. A genetic model for
colorectal tumorigenesis. Cell. 1990;61(5):759-67.
4. Liu Y, Bodner WF. Carcinogênese colorretal. In: Câncer
do Cólon Reto e Ânus. Rossi BM, Nakagawa WT, Ferreira
FO, Aguiar Júnior S. Lopes A, editores. São Paulo.
Lemar-Teccmed, 2004. p.43-54.
5. Wheeler JM, Kim HC, Efstathiou JA, Ilyas M,
Mortensen NJ, Bodmer WF. Hypermethylation of the promotor
region of the E-cadherin gene (CDH1) in sporadic and
ulcerative colitis associated colorectal cancer. Gut. 2003; 48(3):367-71.
6. Wheeler JM, Beck NE, Kim HC, Tomlinson IPM,
Mortensen NJM, Bodmer WF. Mechanisms of inactivation of
mismatch repair genes in human colorectal cancer cell lines:
The predominant role of hMLH1. Proc Natl Acad Sci.
1999; 96(18):10296-301.
7. Wheeler JMD. Epigenetics, mismatch repair and
colorectal cancer. Ann R Coll Surg Engl. 2005;87(1):15-20.
8. Herman J, Umar A, Polyak K, Graff JR, Ahuja N, Issa JPJ,
et al.. Incidence and functional consequences of hMLH1
promoter hypermethylation in colorectal carcinoma. Proc Natl Acad
Sci USA. 1998;95(12): 6870-5.
9. Kámory E, Kolacsek O, Ottó S, Csuka O. hMLH1 and
hMSH2 somatic inactivation mechanisms in sporadic colorectal
cancer patients. Pathol Oncol Res. 2003;9(4):236-41.
10. Seril DN, Liao J, Yang GY, Yang CS. Oxidative stress
and ulcerative colitis-associated carcinogenesis: studies in
humans and animals models. Carcinogenesis. 2003;24(3):353-62.
11. Schmutte C, Yang A S, Nguyen TT, Beart RW, Jones
PA. Mechanisms for the involvement of DNA methylation in
colon carcinogenesis. Cancer Res. 1996;56(10):2375_81.
12. Kondo S, Toyokuni S, Iwasa Y, Tanaka T, Ondera H, Hiai H, et
al. Persistent oxidative stress in human colorectal carcinoma, but
not in adenoma. Free Radic Biol Med. 1999;27(3-4):401-10.
13. Ferreira ALA, Matsubara LS. Radicais livres: conceitos,
doenças relacionadas, sistema de defesa e estresse oxidativo.
Rev Ass Med Bras. 1997;43(1):61-8.
14. Cadenas E, Davies KJ. Mitochondrial free radical
generation, oxidative stress, and aging. Free Rad Biol Med.
2000;29(3-4):222-30.
15. Pravda J. Radical induction theory of ulcerative colitis.
World J Gastroenterol. 2005;11(16);2371-84.
16. Halliwell B, Gutteridge JM. The antioxidants of
human extracellular fluids. Arch Biochem Biophys.1990;280(1):1-8.
17. Gutteridge JM, Halliwell B. Free radicals and antioxidants
in the year 2000. A historical look to the future. Ann N Y
Acad Sci. 2000;899:136-47.
18. Duthie SJ, Collins AR The influence of cell growth,
detoxifying enzymes and DNA repair on hydrogen
peroxide-mediated DNA damage (measured using the comet assay) in
human cells. Free Radical Biol Med. 1997; 22(4):717_24.
19. Pool-Zobel BL, Leucht U. Induction of DNA damage in
human colon cells derived from biopsies by suggested risk factors
of colon cancer. Mutat Res. 1997;375(2):105-16.
20. Ames BN, Shigenaga MK, Hagen TM. Oxidants,
antioxidants, and the degenerative diseases of aging. Proc Natl Acad
Sci USA. 1993;90(17): 7915-22.
21. Ribeiro ML, Priolli DG, Miranda DDC, Arçari DP,
Pedrazzoli Júnior J. Martinez CAR. Analysis of oxidative DNA
damage in patients with colorectal cancer. Clin Colorectal
Cancer. 2008;7(4):267-72.
22. Pinho MSL, Rossi BM. As proteínas envolvidas
na carcinogênese colorretal (IV). Rev bras
Coloproct. 1998;18(4):278-82.
23. Blasi MF, Ventura I, Aquilina G, Degan P, Bertario L, Bassi
C, et al. A human cell-based assay to evaluate the effects
of alterations in the MLH1 mismatch repair gene. Cancer
Res. 2006;66(18):9036-44.
24. Menigatti M, Truninger K, Gebbers JO, Marbet U, Marra
G, Schär P. Normal colorectal mucosa exhibits sex- and
segment-specific susceptibility to DNA methylation at the
hMLH1 and MGMT promoters. Oncogene. 2009;28(6):899-909.
25. Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS,
Houlihan JVPS, et al. MGMT promoter methylation and field defect
in sporadic colorectal cancer. J Nat Cancer Inst. 2005;97(18):1330-8.
26. Oliva MR, Ripoll F, Muniz P, Iradi A, Trullenque R, Valls
V, et al. Genetic alterations and oxidative metabolism in
sporadic colorectal tumors from a Spanish community. Mol
Carcinog. 1997;18(4):232-43.
27. Pool-Zobel BL, Abrahamse SL, Collins AR, Kark W,
Gugler R, Oberreuther D, et al. Analysis of DNA strand
breakes, oxidized bases, and glutathione S-tranferase P1 in human
colon cellsfrom biopsies. Cancer Epidemiol Biomarkers
Prevention. 1999;8(7):609-14.
28. Ladeira MS, Rodrigues MA, Freire-Maia DV, Salvadori
DM. Use of comet assay to access DNA damage in patients
infected by Helicobacter pylori: comparisons between visual and
image analysis. Mutat Res. 2005;586(1):76-86.
29. Buckhaults P, Rago C, St Croix B, Romans KE, Saha S, Zhang
L, et al. Secreted and cell surface genes expressed in benign
and malignant colorectal tumors. Cancer Res. 2001;61(19):6996-7001.
30. Pilcher J. Free radicals. Neonatal Netw. 2002;21(7):33-7.
31. Singh NP, McCoy MT, Tice RR, Schneider EL. A
simple technique for quantitation of low levels of DNA damage
in individual cells. Exp Cell Res. 1988;175(1):184-91.
32. D'Inca R, Cardin R, Benazzato L, Angriman I, Martines
D, Sturniolo GC. Oxidative DNA damage in the mucosa
of ulcerative colitis increases with disease duration and
dysplasia. Inflamm Bowel Dis. 2004;10(1):23-7.
33. Kawanishi S, Hiraku Y, Pinlaor S, Ma N. Oxidative
and nitrative DNA damage in animals and patients
with inflammatory diseases in relation to
inflammation-related carcinogenesis. Biol Chem. 2006;387(4):365-72.
34. Ribeiro ML, Priolli DG, Miranda DDC, Paiva DA,
Pedrazzoli Júnior J, Martinez CAR. Avaliação do dano oxidativo ao
DNA de células normais e neoplásicas da mucosa cólica de
doentes com câncer colorretal. Rev bras Coloproct.
2007;27(4):391-402.
35. Han HJ, Maruyama M, Baba S, Park JG, Nakamura
Y. Genomic structure of human mismatch repair gene,
hMLH1, and its mutation analysis in patients with hereditary
non-polyposis colorectal cancer (HNPCC). Hum Molec
Genet. 1995; 4: 237-42.
36. Kolodner RD, Marsischky GT. Eukaryotic DNA
mismatch repair. Curr Opin Genet Dev. 1999;9(1):89-96.
37. Mazurek A, Berardini M, Fishel R. Activation of human
MutS homologs by 8-oxo-guanine DNA damage. J Biol
Chem. 2002;277(10):8260-6.
38. Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM,
Carter KC, Rosen CA, et al. Mutation of a mutL homolog
in hereditary colon cancer. Science. 1994; 263:1625-9.
39. Sanchez de Abajo A, de la Hoya M, van Puijenbroek M,
Godino J, Díaz-Rubio E, Morreau H, et al. Dual role of LOH at
MMR loci in hereditary non-polyposis colorectal cancer?
Oncogene. 2006;25(14):2124-30.
Endereço para correspondência:
Carlos Augusto Real Martinez
Rua Rui Barbosa, 255 apto. 32
Vila Boa Vista - Santo André - São Paulo
CEP: 09190-370 - Tel: (11) 4438-9203
E-mail: caomartinez@uol.com.br
Recebido em 07/08/2009
Aceito para publicação em 04/09/2009
Trabalho realizado no Programa de Pós-Graduação em Ciências da Saúde da Universidade São Francisco, Bragança Paulista, São Paulo.
Conflito de interesses: nenhum
Fonte de Financiamento: Fundação de Amparo a Pesquisa de Estado de São Paulo (FAPESP). Número do Projeto: nº 08-51499-7.